Key Information
Source
Year
2024
summary/abstract
Renal amyloidosis is a set of complex disorders characterized by the deposition of amyloid proteins in the kidneys, which causes gradual organ damage and potential kidney failure. Recent developments in diagnostic methods, particularly mass spectrometry and proteome profiling, have greatly improved the accuracy of amyloid typing, which is critical for disease management. These technologies provide extensive insights into the specific proteins involved, allowing for more targeted treatment approaches and better patient results. Despite these advances, problems remain, owing to the heterogeneous composition of amyloid proteins and the varying efficacy of treatments based on amyloid type. Access to sophisticated diagnostics and therapy varies greatly, highlighting the global difference in renal amyloidosis management. Future research is needed to investigate next-generation sequencing and gene-editing technologies, like clustered regularly interspaced short palindromic repeats (CRISPR), which promise more profound insights into the genetic basis of amyloidosis.
Keywords:
amyloidosisexternal link, opens in a new tab; immunofluorescenceexternal link, opens in a new tab; immunohistochemistryexternal link, opens in a new tab; mass spectrometryexternal link, opens in a new tab; proteomicsexternal link, opens in a new tab; next-generation sequencingexternal link, opens in a new tab
1. Characterizing Proteomic Variations in Amyloidosis
Amyloidosis is the accumulation of misfolded proteins outside cells in an insoluble β-pleated sheet form. When seen under polarized light and stained with Congo red, these amyloid deposits can be identified by their distinctive apple-green–orange birefringence. Electron microscopy has shown that these deposits comprise rigid, non-branching fibrils measuring 7.5 to 10 nm in diameter [1external link, opens in a new tab,2external link, opens in a new tab]. Localized amyloidosis arises when amyloids are produced and deposited in the same place. In systemic amyloidosis, amyloidogenic proteins are synthesized in one region, such as the bone marrow or liver, and then deposited in another, such as the heart or kidneys [3external link, opens in a new tab]. The mechanisms that cause amyloidosis, whether systemic or localized, are generally classified into three broad categories: (1) the increased production of proteins that result in a predisposition toward amyloid formation, (2) mutations in proteins that increase the likelihood of misfolding compared to their normal counterparts, and (3) a natural proclivity of certain normal (wild-type) proteins to form amyloid [4external link, opens in a new tab]. Systemic amyloidosis can be hereditary or acquired. Notably, the absence of a family history does not preclude hereditary amyloidosis. Approximately half of the people with hereditary amyloidosis have no known family history of the disease [4external link, opens in a new tab,5external link, opens in a new tab,6external link, opens in a new tab].
Amyloidosis is classified based on the originating protein and is typically categorized as either a wild-type (acquired) or germline pathogenic variant (mutant). Proper amyloid typing is vital to avoid diagnostic mistakes [4external link, opens in a new tab,5external link, opens in a new tab,6external link, opens in a new tab]. Forty-two proteins are amyloidogenic, with at least 17 being capable of causing systemic disease [7external link, opens in a new tab]. Presently, six amyloidogenic proteins have the ability to form amyloids in both the wild-type and mutant forms. These proteins include transthyretin (ATTR), beta-2 microglobulin (Aβ2M), serum amyloid A (AA), apolipoprotein A-IV (ApoAIV), Aβ protein (Aβ), and prion protein (APrP) [8external link, opens in a new tab,9external link, opens in a new tab]. Other examples of proteins include leukocyte chemotactic factor 2 (ALECT2), fibrinogen α-chain (AFib), gelsolin (AGel), apolipoproteins A-I and A-II (AApoAI and AApoAII), and apolipoprotein C-III (AApoCIII) [10external link, opens in a new tab]. The subtypes of amyloidosis and their respective pathogenic mechanisms are illustrated
Image
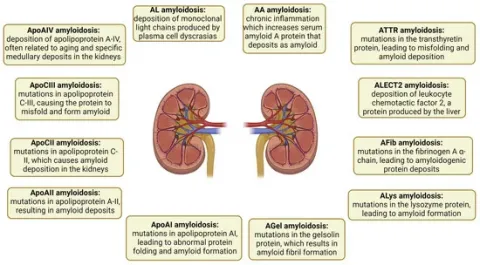
Besides amyloidogenic proteins, multiple other proteins have been identified that contribute to our understanding of the physiology of the disease. Proteins such as clusterin, vitronectin, apolipoprotein E (ApoE), ApoA4, and serum amyloid P-component (SAP) constitute amyloid signature proteins found in all types of amyloidosis and across all affected tissues. While some proteins, such as SAP, may promote amyloid development, others, such as clusterin, may function as amyloid chaperones and reduce amyloid production. Amyloid plaques contain proteins from the affected organs, which can provide information about the degree of amyloid accumulation, the processes that lead to cell damage, and the physiological reactions of various tissues to amyloid accumulation [11external link, opens in a new tab]. Proteins deposited in amyloid plaques (expanded proteome) and proteins overexpressed in plaques compared to controls (plaque-specific proteome) have been investigated in 2650 cases from eight different types of kidney amyloidosis [immunoglobulin light chain (AL), heavy chain (AH), a mix of heavy and light chain amyloid (IGH), ALECT2, AA, AFib, AApoAIV, and AApoCII] using laser microdissection–mass spectrometry. The proteomic content was higher in the more indolent types of amyloid (non-AL) than in AL [12external link, opens in a new tab]. The proteomic “density” of amyloid plaques can serve as an indicator of more gradual disease progression [13external link, opens in a new tab]. AA, AFib, and AApoCII amyloidosis were the most proteomically distinct, driven by increased complement pathway proteins, whereas AL and ALECT2 showed significant proteomic heterogeneity [14external link, opens in a new tab].
Complement pathway proteins are important sources of diversity across various amyloidosis types and proteomic subgroups. They may be synthesized in situ and may signify an unusual response to pathogens. A noticeable reduction in ubiquitins and heat shock proteins in patients with AL, AH, AApoCII, and AFib was noted, which may reflect the overutilization of these proteins by the ubiquitin–proteasome system [14external link, opens in a new tab]. This may also apply to heat shock proteins, a widely recognized cluster of chaperones that counteract amyloid formation [15external link, opens in a new tab]. Ubiquitins and heat shock proteins can be directly captured within aggregates of misfolded proteins, further exacerbating the collapse of cellular proteostasis [16external link, opens in a new tab,17external link, opens in a new tab,18external link, opens in a new tab].
The proteome specific to kidney AL plaques comprised 24 proteins, including those linked to kidney damage (α1 antitrypsin and heat shock protein β1). Hierarchical clustering of AL cases using their plaque-specific proteomes revealed four distinct clusters. One cluster, associated with better kidney survival, was distinguished by its higher proteomic content and the presence of 14-3-3 proteins; yet, it had reduced levels of amyloidogenic light chains (LCs) and the majority of signature proteins. The 14-3-3 proteins are a family of structurally similar phospho-binding proteins that regulate major cellular functions and act as molecular chaperones for several other proteins involved in a variety of cellular processes such as signal transduction, proliferation, differentiation, apoptosis, and autophagy [19external link, opens in a new tab]. They interact directly with different forms of amyloid and help to eliminate them [20external link, opens in a new tab,21external link, opens in a new tab]. Proteins linked to kidney diseases, including heat shock protein family B (small) member 1 (HSPB1), S100A6, transgelin (TAGLN), and TIMP metallopeptidase inhibitor 3 (TIMP3), were shown to be enriched in the proteome unique to AL. HSPB1 enhances autophagy during acute kidney injury (AKI) and is similarly activated by misfolded proteins [22external link, opens in a new tab]. S100A6, a protein that binds calcium, is elevated in AKI and potentially reduces β-amyloid accumulation in animal studies of Alzheimer’s disease [23external link, opens in a new tab,24external link, opens in a new tab]. TAGLN associates with cytoskeletal proteins and is upregulated by the amyloid precursor protein in Alzheimer’s disease [25external link, opens in a new tab]. Lastly, TIMP3, a component also found in cardiac amyloid proteome, may play a role in matrix remodeling and fibrosis in AL [13external link, opens in a new tab]. Glutathione-S transferase (GST) and phosphatidylethanolamine-binding protein 1 (PEB1) showed the highest upregulation in the proteome of early-stage amyloidosis. Both function as antioxidants, and PEB1 plays a key role in regulating the ferroptosis pathway [26external link, opens in a new tab,27external link, opens in a new tab]. Finally, the accumulation behavior of amyloidogenic proteins is independent of other proteins, suggesting that kidney toxicity in this condition cannot be solely attributed to amyloid levels. Investigating the functions of the proteins regarding disease development could provide a foundation for creating new treatments [12external link, opens in a new tab].